


































Molecular details of the ferroptotic program are emerging and one of the key questions is whether the Fe-driven process is enzymatic or based on random free radical reactions. The combined efforts of three HFSP-funded grant teams (Conrad in Munich, Germany; Kagan in Pittsburgh, USA; Klein-Seetharaman in Warwick, UK; see also Greasing up ferroptotic cell death) previously established that hydroperoxy-derivatives of one particular phospholipid, phosphatidylethanolamine (PE), arachidonoyl (C20:4)- or adrenoyl(C22:4)-residues represent pro-ferroptotic signals. Depletion of these PE substrates by genetic ablation of the genes encoding the participating enzymes, acyl-CoA synthetase long chain family member 4 (ACSL4) or lysophosphatidylcholine acyltransferase 3 (LPCAT3), suppresses ferroptosis.
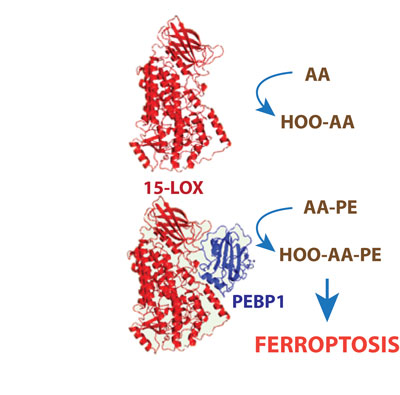
Figure 1: PEBP1 allosterically modifies 15-LOX to permit liganding of larger PE molecules.
Yet, the specificity of the oxidation products implies that the mechanism of their formation includes catalysis of PE oxidation by a non-heme Fe-containing di-oxygenase, 15-lipoxygenase (15-LOX). But how does the enzyme select and recognize specific individual PE molecules among thousands of molecular species of other phospholipids? Moreover, the common substrates for 15-LOX are free polyunsaturated fatty acids, not phospholipids. Then, how does the enzyme accommodate a much bulkier phospholipid substrate within its catalytic site. Recent work from the Kagan laboratory (Cell, October 2017 [1]) addressed some of these questions. It turned out that 15-LOX forms a complex with a scaffold-protein, known as PE-binding protein 1 (PEBP1), that allosterically modifies the enzyme and expands its “catalytic lobby” to permit liganding of larger PE molecules (Fig. 1). Furthermore, PEBP1 binds free fatty acids (at ratios of up-to 9:1), thus depleting the amounts of available PUFA in the vicinity of 15-LOX/PEBP1 complexes. Interestingly, these mechanisms were shown to be enacted in the ferroptotic death of airway epithelial cells in asthma, kidney tubular epithelial cells in acute renal injury and neurons in brain trauma. This suggests that small molecule regulators of 15-LOX/PEBP1 complexes may represent new targets for drug discovery.
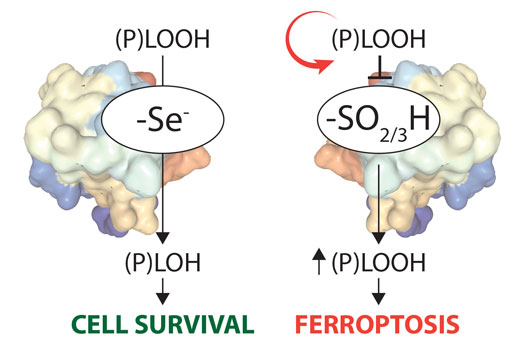
Figure 2: The GPX4-Cys variant is highly sensitive to peroxide-induced, irreversible overoxidation.
The Conrad and Ursini (Padua, Italy) laboratories have recently solved another mystery, why the key ferroptosis regulator GPX4 is a selenoprotein (Cell, December 2017 [2]). Unlike in most organisms that use thiolate-based enzyme catalysis, vertebrate GPX4 uses the 21st amino acid selenocysteine in its active site (Fig. 2). As selenocysteine utilization is a highly complex and energetically costly process, we hypothesized that there must be a true advantage of selenolate- vs thiolate-based catalysis. To this end, a new transgenic mouse line, where the active site selenocysteine was replaced by cysteine, was generated. Unexpectedly, mice homozygous for the GPX4-Cys variant (on a mixed genetic background) developed normally and survived up to three weeks after birth, which is in stark contrast to the early embryonic lethal phenotype observed in GPX4 knockout mice. Yet, homozygous mice displayed severe seizures, therefore they had to be sacrificed. Loss-of a distinct type of inhibitory interneuron, which thus represents the cell type requiring selenium-containing GPX4, was deciphered as the underlying cause of seizures. In-depth mechanistic studies demonstrated that the GPX4-Cys variant is prone to peroxide-induced, irreversible enzyme over-oxidation in vivo, thereby triggering ferroptotic cell death. Hence, it seems that the development of complex neuronal circuits (as evident in vertebrates) requires the exploitation of a rich set of polyunsaturated fatty acids in membranes that comes with the inherently increasing risk of oxidative destruction, while the latter is efficiently counteracted by selenium-containing GPX4.
References
[1] Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Ingold, I., Berndt, C., Schmitt, S., Doll, S., Poschmann, G., Buday, K., Roveri, A., Peng, X., Porto Freitas, F., Seibt, T., et al. (2017). Cell. doi: 10.1016/j.cell.2017.11.048.
[2] PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Wenzel, S.E., Tyurina, Y.Y., Zhao, J., St Croix, C.M., Dar, H.H., Mao, G., Tyurin, V.A., Anthonymuthu, T.S., Kapralov, A.A., Amoscato, A.A., et al. (2017). Cell 171, 628-641 e626.