


































Down syndrome is a common cytogenetic condition seen in newborns and is caused by having an extra copy of chromosome 21, which is known as trisomy 21. In up to 30% of newborns with Down syndrome a transient preleukemia disease occurs. As this preleukemia initiates during fetal development, we characterized the cellular and molecular context of preleukemia using CRISPR/Cas9-mediated gene editing in human disomic and trisomic fetal hematopoietic stem cells and transplantation in mice. Mutations in the erythroid–megakaryocyte transcription factor GATA1 caused transient preleukemia only when introduced into trisomy 21 long-term hematopoietic stem cells, but not when introduced in disomic stem or progenitor cells, likely representing the cell of origin.
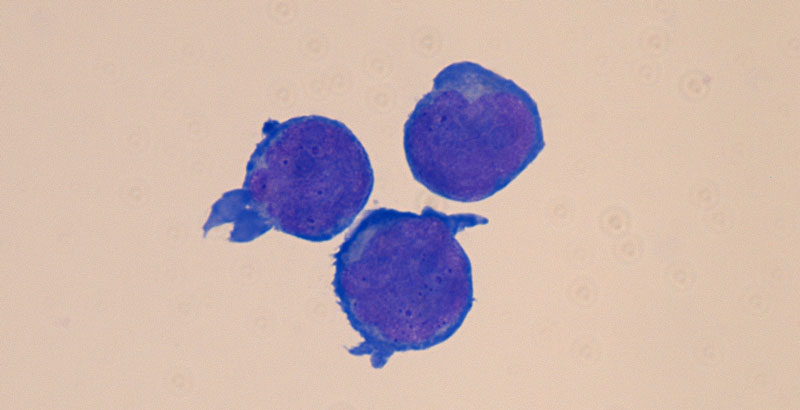
Figure: Preleukemia was initiated upon mutations in GATA1 in trisomy 21 hematopoietic stem cells (Giemsa staining, 100x).
In order to shed light on the reason why an extra copy of chromosome 21 predisposes to leukemia, chromosome 21 miRNAs were identified to be up-regulated in trisomy 21 long-term hematopoietic stem cells compared to disomic 21 stem cells. These miRNAs facilitated predisposition to preleukemia, as knock out of these miRNAs in trisomy 21 long-term hematopoietic stem cells prevented preleukemia development in combination with GATA1 mutations.
Although the transient preleukemia spontaneously disappears within the first months after birth, in up to 20% of individuals, myeloid leukemia later evolves from the preleukemic clone by acquisition of additional genetic mutations. By introducing additional mutations in cohesin genes such as STAG2, progression to leukemia was observed. Surprisingly, this step of leukemia progression was independent of trisomy 21 and was able to originate in various stem and progenitor cells, highlighting opposing trisomy 21 dependencies and distinct cellular origins for the preleukemia initiation and leukemia progression phase.
HFSP funding was instrumental in getting this project started because it allowed us to develop the methodology of using gene editing in primary human hematopoietic stem cells. By applying this methodology to model Down syndrome leukemia, we were able to establish a model that faithfully recapitulated the human disease. Therapeutic targeting of the preleukemic disease using pharmacological inhibitors against KIT reduced the preleukemic burden in this model and in patient derived samples. Therefore, these results potentially reveal a way to prevent the leukemia before it arises, warranting further clinical evaluations.
|
HFSP award information Long-Term Fellowship (LT000601/2016-L) |