


































Extensive exposition of our body to an exogenous protein antigen leads to the production of antibodies that bind it with high affinity and specificity. This process, termed affinity maturation is one of the pillars of our adaptive immune system and the working principle of vaccines. A fundamental question is to determine, given the three-dimensional structure of an antigen, which regions of the protein - termed epitopes - are most likely to be targeted by affinity-matured antibodies. While a priori any epitope on the surface of a protein structure could be targeted, strong preferences are frequently found empirically. For instance, the majority of vaccine or infection-induced antibodies against SARS-CoV-2 target the receptor binding domain of the spike protein, and more specifically the region that binds the ACE2 protein - the entry point of SARS-CoV-2 in human cells.
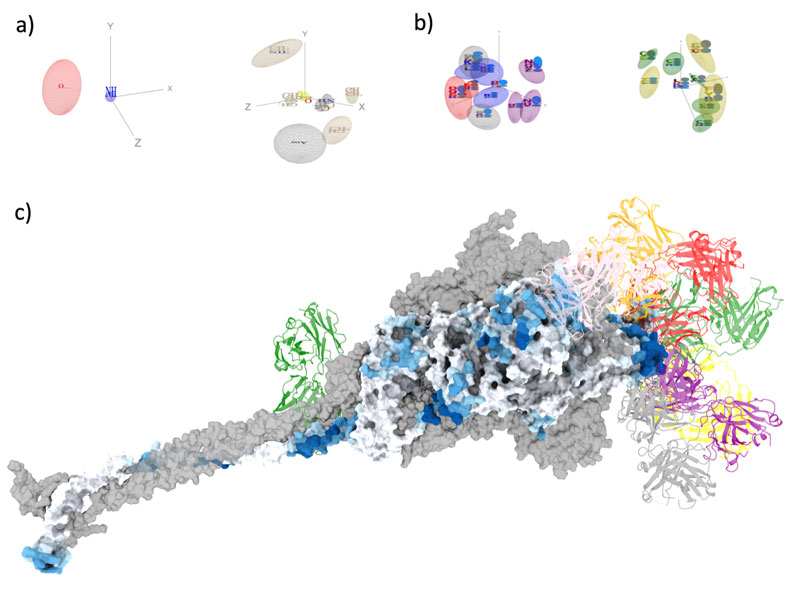
Figure: Illustration of the ScanNet model. a,b) Selected visualizations of spatio-chemical patterns learnt at the atomic (a) and amino acid (b) scale for the prediction of antibody binding sites. Patterns are defined by the presence of specific sets of atoms / amino acids (shown as colored letters with height proportional to their frequency) at prescribed locations (depicted as gaussian ellipsoids). Bottom: Antibody binding site predictions overlaid on the SARS-CoV-2 spike protein trimer (surface representation, colored by probability where white=low and dark blue= high). Representative antibodies are shown in cartoon form.
Systematic mapping of antibody epitopes onto structure is highly challenging. Indeed, unlike, for example, T-cell epitopes, antibody epitopes frequently consist of sets of amino acids that are spatially contiguous but distant along the sequence. Therefore, sequence-based machine learning algorithms often fail to identify them. To tackle this issue, Dr. Jérôme Tubiana, Dr. Dina Schneidman-Duhovny and Pr. Haim J. Wolfson developed ScanNet, an interpretable geometric deep learning model tailored for protein structures. ScanNet extracts local atomic and amino acid neighborhoods within protein structures and passes them through trainable motif-detecting filters. ScanNet was trained to predict epitopes using publicly available experimental structures of antibody-antigen complexes and was found to be significantly more accurate than prior approaches based on comparative modeling or feature-based machine learning. In particular, ScanNet could accurately predict the epitopes of the SARS-CoV-2 spike protein (Fig. 1). Additionally, the filters learned by the network could be readily visualized and interpreted: simple patterns such as hydrogen bonds, secondary structure elements, and exposed hydrophobic residues were found, as well as more complex ones such as hotspots “O-rings” (Fig. 1). The complex representation learned was found to correlate with many known physio-chemical features such as solvent accessibility or electrostatic potential. Taken together, these findings suggest that ScanNet successfully learned some of the fundamental physico-chemical principles underlying antigen recognition.
In December 2021, Omicron, a new SARS-CoV-2 variant of concern emerged, featuring an unprecedented number of mutations and considerable immune escape. This prompted the researchers to investigate possible changes in the antibody epitope profile of Omicron using ScanNet. They found that the epitope profile of the Omicron receptor binding domain is distinct from the prior variants of concern. Indeed, it features reduced epitope propensity on its ACE2 binding site, which is precisely the region most targeted by antibodies. Thus, it is harder for antibodies to bind and subsequently neutralize Omicron than other strains. To substantiate the prediction, the authors collaborated with the experimental research groups of Dr. Yi Shi and Dr. Kong Chen from the University of Pittsburgh. Mice were immunized with different recombinant receptor binding domain variants, and it was found that the serologic response of Omicron was drastically attenuated. These findings corroborated other studies reporting that Omicron breakthrough cases were associated with substantially lower antibody titers than other variants of concern cases.
In the future, ScanNet could prove useful for monitoring the emergence of new variants as antigenicity could further decrease in the coming years. Another potential of ScanNet is the design of non-immunogenic therapeutic proteins; by predicting overall antigenicity levels, ScanNet could facilitate the identification of candidate proteins at high risk of inducing an adverse immune response. The long-term financial support of the HFSP Cross-Disciplinary Fellowship program was determinant for completion of this series of interdisciplinary works.
|
HFSP award information Cross-Disciplinary Fellowship (LT001058/2019-C ): Modelling the sequence - structure - function relationship in proteins with machine learning Fellow: Jérôme Tubiana |