


































Dysfunction in RNA metabolism has emerged as a pivotal mechanism underlying several neurodegenerative diseases, in particular amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), two age-dependent neurodegenerative disorders that are linked mechanistically and clinically. A landmark contribution to understanding cellular mechanisms of ALS and FTD came from the discovery of cytoplasmic accumulation and nuclear loss of the RNA binding protein TDP-43 from affected neurons in most instances of ALS, as well as in approximately half of patients with FTD. TDP-43 plays central roles in RNA metabolism, and it is anticipated that loss of nuclear TDP-43 in affected neurons drives processing alterations of multiple RNAs. Unexplained, however, is how TDP-43 dysfunction is directly linked to provoking degeneration of motor neurons, the key event in development of paralysis in ALS.
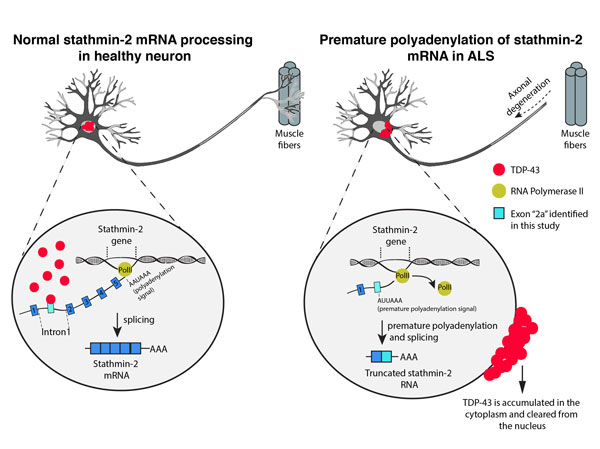
Figure: In healthy neurons (left) TDP-43 is localized to the nucleus and maintains normal processing of stathmin-2 pre-mRNA through binding to intron 1 of the nascent RNA transcript and repression of premature polyadenylation. In ALS (right) TDP-43 is cleared from the nucleus of affected motor neurons, a premature polyadenylation signal (AUUAAA) is uncovered in intron 1 of stathmin-2 pre-mRNA, leading to altered processing and expression of a truncated stathmin-2 RNA. The consequence from suppression of stathmin-2 expression in affected motor neurons is increased vulnerability due to inhibition of axonal regeneration.
By modelling the consequences of TDP-43 loss or mutation in cultured human neurons, we identified a new critical role for TDP-43 in the maturation of mRNAs encoding stathmin-2, a tubulin-binding protein previously implicated in axonal growth and regeneration. We show that normally, TDP-43 binds stathmin-2 transcript to maintain its proper processing into mature mRNA, whereas loss of TDP-43 leads to suppression of stathmin-2 levels as a consequence of truncating the mRNA encoding stathmin-2. Mechanistically, a premature polyadenylation site in intron 1 of stathmin-2 pre-mRNA is uncovered upon TDP-43 loss of function, leading to production of a shortened, aberrant RNA. The direct consequence of this event is nearly complete loss of the stathmin-2 protein product.
Importantly, analysis of post-mortem samples of human spinal cords has identified that aberrant processing of stathmin-2 pre-mRNA is a hallmark of ALS which is consistently found in spinal motor neurons and motor cortex of both sporadic ALS and familial ALS patients carrying a repeat expansion in the C9orf72 gene, the most frequent genetic cause of ALS and FTD.
Stathmin-2 has been associated with neuronal growth and regeneration, most likely due its function in microtubule dynamics. To test whether suppression of stathmin-2 increases motor neuron vulnerability, we cultured human motor neurons derived from induced pluripotent cells (iPSCs) in a two-compartment microfluidic device. Application of mechanical damage to their axonal processes in the distal compartment (without affecting the neuronal bodies) led to rapid axonal regeneration accompanied by accumulation of stathmin-2 in the growth cones and regenerating axons. Reduction in either TDP-43 or stathmin-2 almost completely suppressed motor neuron regeneration after initial damage. Remarkably, although loss of TDP-43 affects the levels or splicing of 1500 mRNAs, restoration of stathmin-2 to TDP-43 depleted motor neurons restored axonal regeneration capacity, strongly supporting stathmin-2 as an essential component of motor neuron regeneration.
In summary, our study identified premature polyadenylation-mediated suppression of stathmin-2 as a new hallmark of ALS/FTD that functionally links reduced nuclear TDP-43 function to enhanced neuronal vulnerability. These findings highlight probable dysfunction in microtubule dynamics as a pivotal mechanism underlying ALS and seed the basis for future therapeutic approaches restoring stathmin-2 levels in multiple neurodegenerative diseases in which TDP-43 loss is known to be a prominent pathology.
The HFSP Long-Term Fellowship was instrumental for the transition from basic RNA research into studying neurodegeneration and ALS, providing the inspiration and confidence that by moving from one’s comfort zone – an unexpected discovery may arise.