


































Have you ever thought about how the world may have looked 250,000 generations ago? If we had a time machine, just like HG Wells in his famous novel ‘The Time Machine’ we might find out. On a human time scale, that would lead us to our ancestors whom we share with apes, but for bacteria this means travelling back just 70 or so years. That is about when humans started to develop chemical substances, called phosphotriesters, which were supposed to kill creepy creatures. And, not too surprisingly, considering human nature, they were used as warfare agents too. Some of these xenobiotics accumulated in the soil around the factories they were produced in. There, 1000s of different bacteria lived and thrived. And just some of them (Pseudomonas) learned, slowly but steadily, to make use of these xenobiotics from chemical industry.
But just HOW did these bacteria achieve this? We do not know. No, halt! We DO know – now! Thanks to elaborate biochemical lab work and new methods from bioinformatics, we can find out! An HFSP funded team, led by Prof. Nobuhiko Tokuriki (Vancouver) who managed the lab work, and Prof. Erich Bornberg-Bauer (Münster, PI on the HFSP grant) who supervised the computational reconstruction of ancestral enzymes (carried out by Elias Dohmen), DID find out.
The researchers started off with extant enzymes (phosphotriesterases), proteins found in today’s bacteria, to reconstruct how these proteins had changed, i.e. mutated, over the last 70 years. The proteins have some 300 amino acids, each drawn from a set of 20 different such building blocks. With modern bioinformatics methods, so called ancestral sequence reconstruction based on statistical methods (Maximum Likelihood), they could reconstruct the most likely ancestral enzymes. This was the basis to also reconstruct the most likely evolutionary path, that is the order in which the mutations may have occurred since then, to yield the extant enzymes.
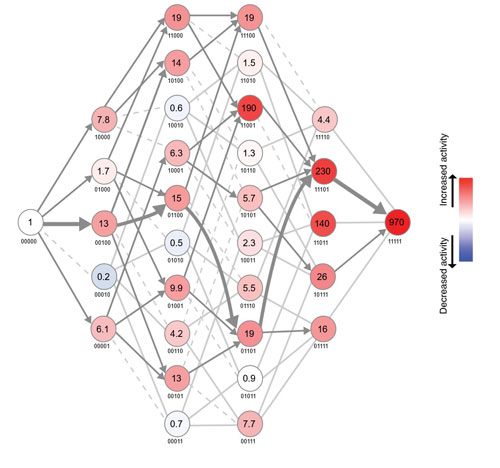
Figure: The adaptive landscape between the likely reconstructed ancestor (left) and the extant wild type (right) for activity of MPH. Each node represents a unique variant, with the genetic background indicated where ‘0’ refers to the ancestral and ‘1’ refers to the derived state at that position (for example, 10000 denotes the ancestral enzyme with just the first position along the sequence mutated from Leu (ancestral state) to Arg (extant)). Numbers in each node indicate the activity increase relative to the ancestor (see colour code on the right). Edges connecting the nodes correspond to point mutations: dark grey arrows indicate evolutionary accessible mutations as they do not decrease the activity (fitness), solid light grey lines do not decrease fitness but are not accessible because all paths leading to the previous node were already inaccessible, dashed light grey lines are inaccessible paths due to a decrease in fitness.
The team found there were some 32 positions (out of the 300) which had changed over time. Trying them all out would amount to 232 (~4 × 109) sequences (including intermediate mutations which we cannot reconstruct anymore it would be 3220 ~ 1030) -- way too many to be accomplished even with all the HFSP grants awarded to date! However, 5 mutations stood out and the combination of amino acids found in these positions reduced the number of mutant proteins to be tested for their activities to 25 (32).
Still a daunting task for a single project, but Gloria Yang from the Tokuriki lab managed it perfectly! It was, therefore, possible to analyse all paths, i.e. all possible sequential orders of all 5 mutations and the effects which all combinations of 2, 3, 4 and all 5 of the mutations had together, so called 'epistatic interactions'.
Most of the paths got stuck at an intermediate stage, with the effectivity of the enzyme in degrading the xenobiotic substrate decreased. This would have prevented further evolution but it turned out that a fraction of the 120 possible paths were ‘smooth’ enough, with all steps going more or less ‘up’ (becoming ‘fitter’), from the very inefficient ancestral state to the very efficient extant enzyme. The researchers identified one particular path that’s believed to be the most likely, or close to the most likely path which natural evolution has taken.
Results were further corroborated by molecular dynamics studies carried out by the team of Colin Jackson in Canberra, Australia.
Eventually, the researchers had a very clear picture of how such ‘higher order epistatic pathways’ work. And it became clear how, from one enzyme which originally digested a naturally occurring substance called dihydrocoumarin (also used as an additive in foods, as well as a fragrance in cosmetics) learned to live on poison. Apart from explaining how specialisation of a promiscuous enzyme (a multi-functional enzyme acting on several different substrates) worked, this study has also ramifications for developing new strategies for directed evolutionary protein design, a field which was highlighted by the 2018 Nobel prize in Chemistry, awarded to Prof. Frances Arnold (Caltech).
If only HG Wells had known all this! Would he have changed his story of time travel? We don't know, but maybe, one day, humans too can travel in time, back to the late 19th century, straight into the writing room of HG Wells, perhaps secretly altering his manuscripts before typesetting. Maybe, maybe ...