


































Melanin, the pigment that colors our skin, hair, and eyes, is produced and stored inside specialized compartments within cells. A key step in this process is the formation of a nanoscale protein scaffold, made from the protein PMEL, which provides a surface on which melanin can accumulate. Although PMEL belongs to the broader family of “amyloid” proteins—often associated with diseases such as Alzheimer’s—PMEL is an example of a functional amyloid that is essential for normal biology. Until recently, the precise three-dimensional structure of these physiological amyloid fibers had not been visualized at atomic resolution.
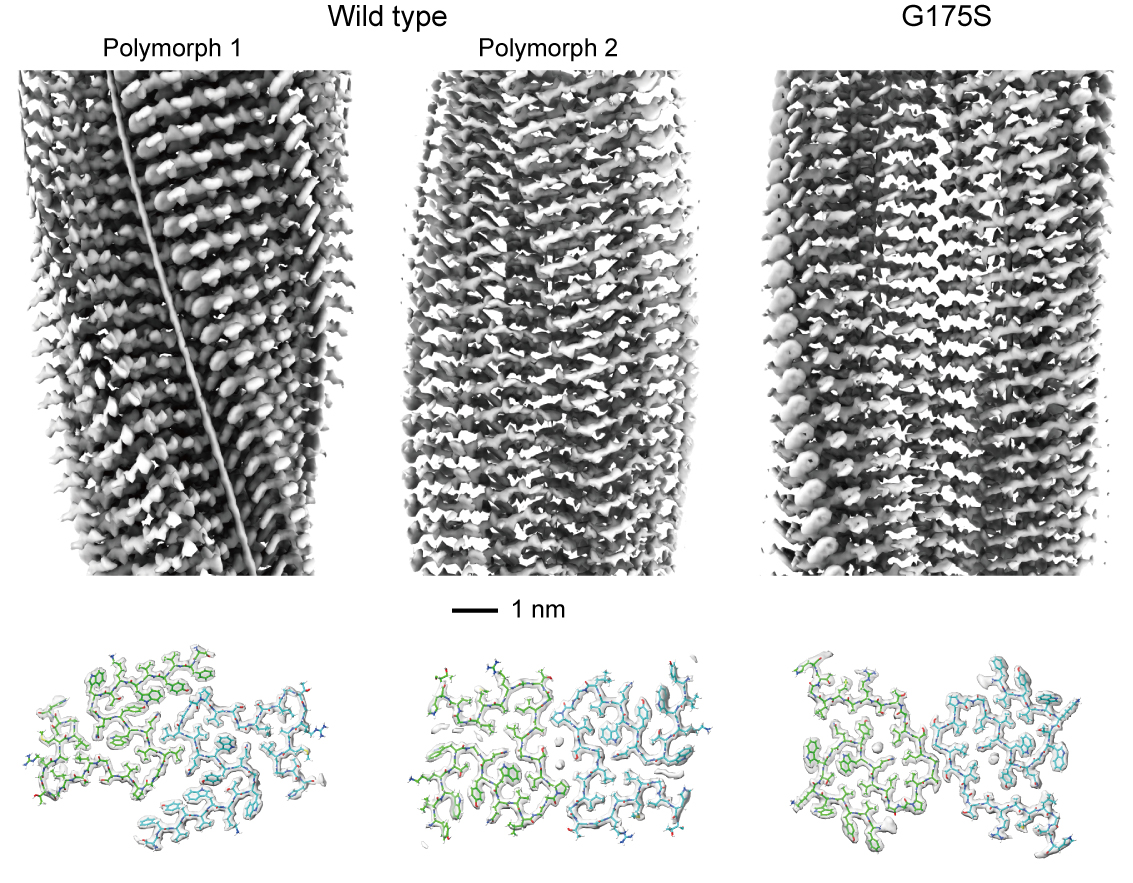
Using cryo-electron microscopy, the HFSP Research team determined the near-atomic structures of PMEL amyloid cores purified directly from human pigment cells. They discovered that the PMEL scaffold naturally adopts two distinct structural forms, or polymorphs, each built from paired protein filaments. Despite their similar β-sheet compositions, these scaffolds differ strikingly in shape, packing density, and the presence or absence of small internal cavities. This structural diversity may help pigment cells organize melanin efficiently within their storage compartments.
The scientists next examined the PMEL variant Gly175Ser, a single-amino-acid mutation strongly associated with pigment dispersion syndrome (PDS), a condition in which pigment escapes from the iris and can block fluid drainage in the eye. This blockage may ultimately lead to pigmentary glaucoma. The structural analysis revealed that this mutation introduces a new hydrogen bond that stabilizes an alternative amyloid conformation, subtly but decisively reshaping the scaffold. Consistent with this structural change, the mutant protein formed amyloids four times more efficiently in vitro and produced substantially increased amyloid loads inside and outside living cells.
Despite the excess amyloid, the overall architecture of melanin-storing organelles (melanosomes) remained intact, as shown by cryo-electron tomography. However, cells expressing the mutant protein contained more melanosomes at the stage where melanin begins to deposit, indicating that the altered scaffold may accelerate or dysregulate pigment-assembly steps. Together, these findings suggest a molecular mechanism in which a hyper-stable PMEL scaffold leads to abnormal pigment release, providing a structural explanation for how PDS may begin.
This study sheds light on how a tiny structural change, down to a single bond, can cascade into altered pigment handling in the eye. More broadly, it illustrates how functional amyloids differ from pathological ones and how their precise architectures determine biological outcomes. These insights could guide future development of diagnostic approaches or therapeutic strategies for pigmentary glaucoma and may help refine our understanding of amyloid behavior across biological systems.