


































While all cells in our body share the same genome, different cells types employ different genomic regulatory elements to drive the expression of distinct sets of genes, conferring their distinct cell states and phenotypes. These differential regulatory epigenetic states are specified by differential chromatin accessibility and DNA and chromatin modifications (the word "epigenetics’’ is of Greek origin and literally means "over and above’’ the genome). DNA accessibility is central to epigenetic regulation as it affects the ability of different molecules to physically access DNA; active gene promoters and distal regulatory elements are typically characterized by accessible DNA, to which the transcriptional machinery and regulatory proteins can bind. In turn, mapping the DNA accessibility landscape of a cell is key to understanding its regulatory state.
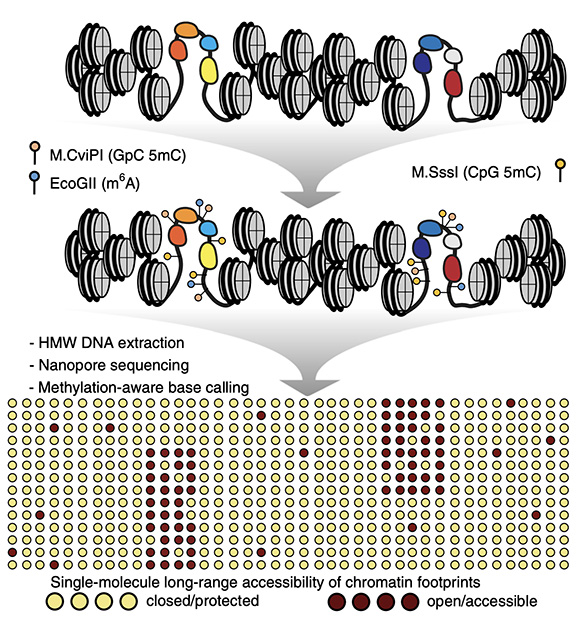
Figure 1: Graphical abstract showing the steps taken to probe DNA accessibility using long DNA molecules. Live cells are permeabilized and 3 methyltransferase enzymes are added to the cells, the enzymes methylate the DNA at regions of DNA accessibility while being blocked from DNA regions that are not accessible. The outcome of the process is an accessibility landscape across long molecules of DNA.
Measurements of DNA accessibility take advantage of the preferential action of various enzymes and small molecules on accessible DNA versus inaccessible DNA. The most well-known and widely used methods are DNase-seq and ATAC-seq. Both methods rely on the ability of different enzymes (DNAse I and the Tn5 transposase, respectively) to cleave DNA within accessible chromatin. A sequencing library consisting of short DNA fragments is then prepared and sequenced, after which accessible regions are identified genome-wide. However, because DNA is fragmented, accessible DNA is enriched, and short reads are sequenced, such measurements can only map localized regions of statistical enrichment and are unable to provide key pieces of information:
First, we are unable to quantitatively measure the distribution of regulatory states within a population of cells, i.e., how many cells are active in a given cRE. Second, the joint regulatory state of multiple elements along the chromatin fiber cannot be measured, yet knowing it is in fact of major importance as transcription of genes is often driven by the action of cREs located at a significant genomic distance.
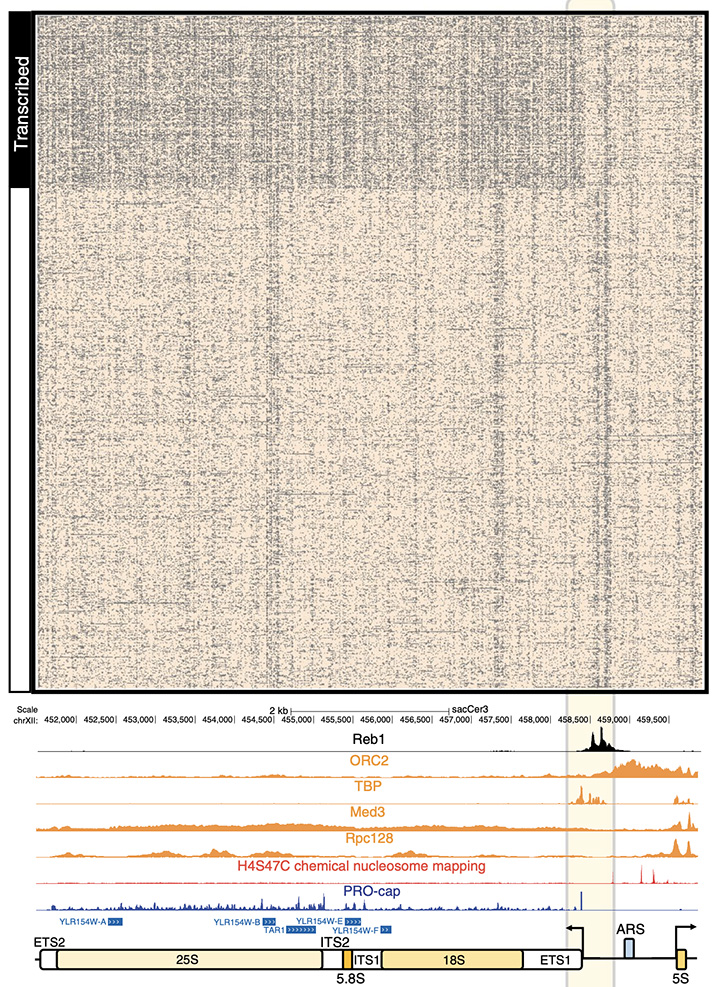
Figure 2: Ribosomal DNA in the Yeast genome is made up of two genes, 35S (made from 18S and 25S) and 5S, transcribing to opposing directions. This unit of two genes exists as an array of roughly 150 repeats to allow for high expression of the ribosomal proteins. Using SMAC-seq we were able to find that only about 50% of the 150 units transcribe genes and that there is only one small regulatory difference between the active to inactive units based on Reb1 regulation.
To overcome the shortcomings of current DNA accessibility techniques and gain new insights into genomic regulation, we developed a single-molecule long-read accessible chromatin mapping sequencing (SMAC-seq) technique. SMAC-seq is a method that directly assays both open chromatin regions and nucleosome positioning within a single chromatin fiber at multikilobase scales. We used SMAC-seq [1] to study chromatin architecture and co-accessibility states in the yeast Saccharomyces cerevisiae. We assessed the degree of coordination between positions of nearby nucleosome particles, enumerated mutually exclusive regulatory states along individual loci and observed coordinated changes in nucleosome positioning and chromatin accessibility upon transcriptional activation. SMAC-seq also allows for footprinting of the occupancy of regulatory proteins, and provides strand-specific information about the exposure of DNA occupied by nucleosomes.